原子尺度的计算模拟对于理解和预测材料微观结构与宏观性能之间的关系至关重要,但要同时实现模拟的准确性和计算效率却颇具难度。基于密度泛函理论的第一性原理计算能够精确计算材料的微观结构特征,但需要较高的时间成本;分子动力学模拟计算效率高,但无法保证结果的精度及准确性。随着计算技术的迅速发展,基于数据驱动的机器学习原子间势能函数已成为材料科学研究的重要工具。该方法可以从实验或高精度的第一性原理计算中获得的大量数据中学习,构建出能够准确描述材料原子间相互作用的机器学习势函数,并将其应用于大规模系统的分子动力学模拟。机器学习势函数平衡了计算效率和精度,能够准确预测材料的物理和化学性质,在材料科学研究领域展现出巨大潜力。论文介绍了机器学习原子间势能的基本构建思路和主要构建方法,综述了其在材料结构预测和材料性质研究中的最新进展,并总结了机器学习势能所面临的挑战和未来的发展方向。
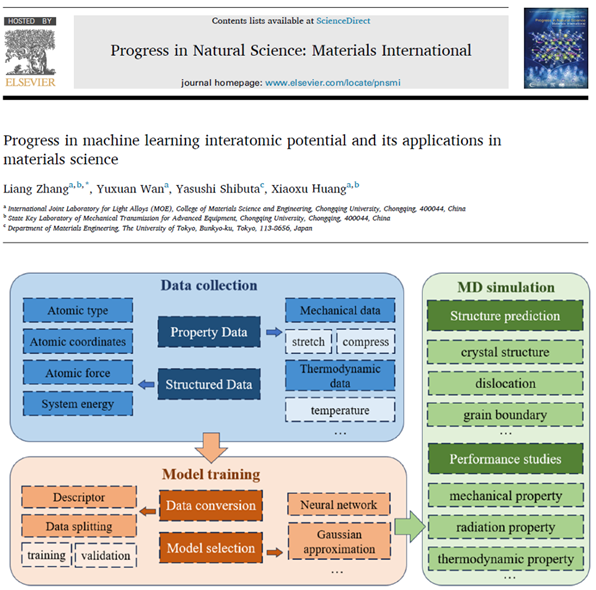
相关工作以题为“Progress in machine learning interatomic potential and its applications in materials science”的综述论文发表在《Progress in Natural Science: Materials International》上,论文作者包括硕士生万雨萱、张亮副教授、黄晓旭教授和日本东京大学Yasushi Shibuta教授。该研究得到了国家自然科学基金面上项目(52571004)和重点项目(52130107)的资助。
论文链接:https://doi.org/10.1016/j.pnsc.2025.11.002